论文总字数:26285字
目 录
摘要 I
Abstract II
引言 1
第一章 综述 1
1.1 红外光谱简介 1
1.2 拉曼光谱简介 2
第二章 计算方法 2
2.1 Gaussian简介 2
2.2 Gaussview简介 2
2.3 密度泛函理论简介 2
2.4 几何优化简介 2
2.5 计算方法 3
第三章 呋喃的红外光谱及拉曼光谱研究 3
3.1 呋喃的几何构型 3
3.2 呋喃分子的红外光谱研究 5
3.3 呋喃分子的拉曼光谱研究 7
第四章 2-甲基呋喃的红外光谱及拉曼光谱研究 9
4.1 2-甲基呋喃的几何构型 9
4.2 2-甲基呋喃的红外光谱研究 10
4.3 2-甲基呋喃的拉曼光谱研究 13
第五章 2,5-二甲基呋喃的红外光谱和拉曼光谱研究 15
5.1 2,5-二甲基呋喃分子的几何构型 15
5.2 2,5-二甲基呋喃的红外光谱研究 17
5.3 2,5-二甲基呋喃的拉曼光谱研究 20
第六章 总结 23
参考文献 23
发表论文目录 25
致谢 26
创新点
关于呋喃类生物质燃料以往是通过实验分析其化学燃烧反应,不同与此,本文利用Gaussian09软件对呋喃、2-甲基呋喃及2,5-二甲基呋喃进行了大量关于构型、光谱计算。 本文成果将为生物质燃料研究领域提供可靠的理论数据。具体如下:
1. 对呋喃分子进行了键长、键角特性研究,分析得到了呋喃分子的解离机理。
2. 得到了2-甲基呋喃分子的红外光谱,有利于日后的物质特征光谱检测,提高了这几种物质的检测效率。
3. 同时,也通过软件计算得到了2,5甲基呋喃分子的拉曼光谱图,便于对此物质的特征吸收光谱的检测。
4. 得到了2-甲基呋喃分子和2,5-二甲基呋喃分子的分子电荷分布对于日后外加电场的研究有着重要的比对作用。
呋喃及其衍生物的光谱研究
张巍
Abstract: In this paper, the density functional theory in Gaussian09 software is used to optimize the furan (C4H4O) and 2-methylfuran (C5H6O) and 2,5 methylfuran (C6H9O) at the B3LYP / 6-311 G (d, p) basis set level. After calculation, the bond length and bond angle of furan and its two derivatives were obtained and compared. In addition, the data of molecular charge distribution were obtained, and the dissociation mechanism was analyzed synthetically. Secondly, the Raman spectra and infrared vibrational spectra of furan and its derivatives were obtained, and the spectra were summarized. Based on the analysis of various factors, we obtained the important physical properties of furan and its derivatives, 2-methylfuran and 2,5-dimethylfuran.
Key words: Furan, 2-methylfuran, 2,5-dimethylfuran, density functional theory, infrared spectroscopy, Raman spectroscopy
引言
能源枯竭和环境恶化是当今世界上各个国家面临的共同挑战。目前石油基燃料仍是主要燃料,燃料给予人们光和热,支撑着人们的衣食住行,涉及到社会的各行各业的正常运作。但石油资源蕴藏量在日益枯竭,其价格也经常大幅度波动,最应该引起重视的是,石油基燃料加速了全球气候变暖。而生物质燃料是可以替代石油基能源的,它是具有巨大潜力的可再生资源,其具有的优势包括:缓解能源短缺问题,保证能源安全,保护生态环境等。所以,寻找可再生的生物质基燃料来代替石油基能源是科学界与工业界的研究热点,也是社会可持续发展及政府与公众高度关注的焦点问题。
呋喃是最简单的含氧五元杂环化合物,其同系物2-甲基呋喃及2,5-二甲基呋喃(DMF)具有高沸点、高能量密度、高辛烷值和不溶于水等特点[1],被科学家们认为是最有前景的液体燃料。国内外自2010年起,先后对呋喃及其衍生物开展了研究。美国宾夕法尼亚州研究基金会于2010年12月14日宣布,由Weiran Yang和Ayusman Sen等开发的一步催化工艺专利,可用于使生物质和生物质衍生的碳水化合物转化为液体呋喃燃料。该专利为将木质纤维素生物质以及生物质衍生的碳水化合物转化为用于制取燃料的四氢呋喃类型化合物方法。[2]这些燃料包括2,5-二甲基呋喃(DMF)等。国内天津大学的“呋喃类生物质燃料的燃烧化学研究”项目,分析呋喃类燃料燃烧过程中的化学反应,构建其燃烧反应机理,为这类燃料的实际使用提供实验数据和理论指导。
本文将研究呋喃及其衍生物分子构型信息、主要解离离子的拉曼、红外振动光谱,得到了该分子的重要信息。对其衍生物进行了红外光谱的分析统计,对于2-甲基呋喃和2,5-二甲基呋喃的解离机制有着重要的意义。因此本文较为系统的陈述研究成果,这将为该类物质今后作为新型液体能源的开发研究提供重要的理论依据,具有相当的应用价值和指导意义。
首先,利用Gaussian09软件包中设置B3LYP/6-311 G(d ,p)方法对呋喃分子进行构型的优化以及红外光谱的计算。根据计算数据进行分析比较,得到了呋喃分子的键长、键角构型参数,以及红外振动光谱。随后利用同样的机组对其衍生物2-甲基呋喃和2,5-二甲基呋喃进行相同的计算,同样得到了键长、键角、构型参数[3]。此外,本文还通过gaussian09计算得到呋喃及其衍生物的主要解离离子的拉曼、红外振动光谱,得到了该分子的重要信息。
最后,我们对呋喃及其衍生物的键长、键角、拉曼、红外振动光谱参数在origin软件中进行了统计,得到呋喃的构型参数的具体规律。
第一章 综述
- 红外光谱简介
红外光谱是研究分子结构和分子振动光谱的一种方式。它的具体操作方式是用一束具有连续波长的红光去照射一种物质,这种物质的每一个分子都会吸收其中一部分光能,并把它变为另外一种能量,即分子的振动能量与转动能量。所以用单色器对通过这种物质之后的光进行色散,就可得到一条谱带,给谱带确定好坐标后即得到红外光谱。
- 拉曼光谱简介
拉曼光谱分析法是基于拉曼散射效应的一种分析方法。拉曼效应指的是,光照射到物质上会发生弹性散射和非弹性散射,弹性散射产生的散射光成分与激发光波长相同,非弹性散射光有比激发光波长短和长的成分,称为拉曼效应。拉曼效应是光子与光学支声子相互作用的结果。拉曼光谱分析法可以分析得到分子的振动、转动方面的信息,也应用于分子结构的研究[4]。
第二章 计算方法
2.1 Gaussian简介
高斯是一个强大的量子化学软件包。 可执行程序可以在不同型号的大型计算机,超级计算机,工作站和个人计算机上运行,并且具有不同的版本。 高斯的功能有:过渡态能量和结构,键和反应能,分子轨道,原子电荷和电位,振动频率,红外和拉曼光谱[5],核磁性,极化性和超极化性,热力学性质,反应路径,计算可以对基态或兴奋激发态的系统。 可以预测周期性系统的能量,结构和分子轨道。 因此,高斯可以作为研究许多化学领域的有力工具,如取代基的影响,化学反应机理,势能面和激发能等, 经常和gaussview一起使用[6]。在长期的计算与研究中发现,在密度泛函B3LYP理论下的6-311G (d, p)基组,对分子结构优化、红外光谱以及拉曼光谱的计算较为精确[3]。
2.2 Gaussview简介
Gaussview是专门设计的一个与Gaussian搭配使用的软件,它有两个主要的用途,其一,是构建Gaussian的输入文件;其二,是将Gaussian的计算结果以图片的形式显示出来。Gaussview除了可以通过自己来构建输入文件以外,还可以读入许多其他的不同格式的文件,可以和许多的图形软件一起使用,有很大的使用范围[7]。
2.3 密度泛函理论简介
密度泛函理论(DFT)是一种基于电荷密度自洽的原理来研究多电子体系当中的电子结构的方法,可以根据所采用的相关或者交换泛函来选择不同的DET方法,这当中B3LYP方法是目前最广泛使用的DFT方法,这是因为DFT方法与HF相比,考虑了电子之间存在的相关作用,因此其得到的能量会更加精确[8]。目前量子化学的计算方法中最常用的就是密度泛函理论,其唯一的缺点是优化过后所得到的键长通常都会偏长[9]。
2.4 几何优化简介
当分子的分子量比较大、原子数比较多的时候,它就会有多维的势能面,势能面是随着分子的几何构型的变化而能量变化的曲线,分子的自由度决定维数,势能面的中间会有一些特殊的点,像全局最大(小)值、局域极大(小)值和鞍点等[10]。几何优化从我们最开始构建的构型开始,计算梯度和能量来决定我们下一步的方向,这个方向总是以能量下降最快为标准,其中有一些优化也会计算能量的二阶导数。怎么样来确定这个构型是不是稳定的,必须满足四个收敛标准:第一,均方根<0.0003;第二,力的最大值< 0.00045;第三,为后面一步做的取代计算< 0.0018;第四,均方根<0.0012。优化过程有的时候会需要很长的时间,比如说体系已经非常地松弛或者力的值已经小于域值,这个时候可以认为系统已经达到了最优点。
2.5 计算方法
首先用Gaussview构造初始构型,然后在HF的3-21G水平上来对构型进行一个初步的优化,把这个优化结果作为初始结构,再在DFT的B3LYP在6-311G (d,p)水平上来进行精确的优化计算[11-15]。
第三章 呋喃的红外光谱及拉曼光谱研究
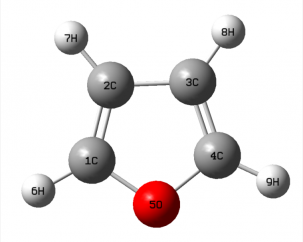
3.1 呋喃的几何构型
在Gaussview中构造呋喃的初始构型,通过HF中3-21G基组水平上对呋喃的构型进行初步优化,再通过密度泛函理论DFT中B3LYP方法在6-311G (d,p)高精度基组上对呋喃的构型进行精确优化计算。
剩余内容已隐藏,请支付后下载全文,论文总字数:26285字
相关图片展示:




该课题毕业论文、开题报告、外文翻译、程序设计、图纸设计等资料可联系客服协助查找;

